Transition Elements
Introduction
Across the second and third periods of the Periodic Table, there is a gradation in properties, from the alkali metals to the halogens. The fourth period begins in the same way, with an alkali metal (potassium) and an alkaline earth (calcium). The next ten elements do not show the gradation in properties of previous periods: they are remarkably similar to one another in their properties and are all metals. They are called the first transition series. Periods five and six also contain transition series.
|
Transition Series |
d subshell being filled |
Elements |
|
First |
3d |
Sc- Zn |
|
Second |
4d |
Y- Cd |
|
Third |
5d |
La- Hg |
The reason for the similarity of the first transition series is that, considering the series from left to right, while each additional electron is entering the 3d shell, the chemistry of the elements continues to be determined largely by the 4s electrons. From one transition element to the next, the nuclear charge increases by 1 unit, and the number of electrons also increases by 1. Since each additional electron enters the 3d shell, it helps to shield the 4s electrons from the increased nuclear charge, with the result that the effective nuclear charge remains fairly constant across the series of transition elements. The sizes of the atoms and the magnitudes of the first ionisation energies are therefore very similar and the elements have comparable electropositivities.
The electron configurations of the first transition series are:
|
Sc |
1s22s22p63s23p64s23d1 |
[Ar]4s23d1 |
|
Ti |
1s22s22p63s23p64s23d2 |
[Ar]4s23d2 |
|
V |
1s22s22p63s23p64s23d3 |
[Ar]4s23d3 |
|
Cr |
1s22s22p63s23p64s13d5 |
[Ar]4s13d5 |
|
Mn |
1s22s22p63s23p64s23d5 |
[Ar]4s23d5 |
|
Fe |
1s22s22p63s23p64s23d6 |
[Ar]4s23d6 |
|
Co |
1s22s22p63s23p64s23d7 |
[Ar]4s23d7 |
|
Ni |
1s22s22p63s23p64s23d8 |
[Ar]4s23d8 |
|
Cu |
1s22s22p63s23p64s13d10 |
[Ar]4s13d10 |
|
Zn |
1s22s22p63s23p64s23d10 |
[Ar]4s23d10 |
You will notice that chromium completes occupation of its d orbitals with unpaired electrons at the expense of its 4s electrons, and copper completes its full d10 shell at the expense of its 4s electrons. There appears to be a certain measure of stability associated with a full d10 shell and with a half-filled d5 shell.
Definition
of a transition element:
Transition elements are often referred to as d-block metals. They are defined as elements which form compounds in which there is an incomplete subshell of d electrons. Scandium (3d0 in compounds) and zinc (3d10 in compounds) are excluded by this definition, and copper is only included in copper(II) (3d9) compounds. It is convenient to include these metals in a treatment of transition elements, however, on account of the chemical resemblance of their compounds to transition element compounds.
The characteristic properties of transition elements arise from the incomplete d subshell in either the atoms or the ions they form. These characteristics include the ability to:
· form compounds with the transition element in variable oxidation states
· form complex ions
· form coloured ions
· act as catalysts
· exhibit paramagnetism.
Variable Oxidation States
One of the characteristic properties of the transition elements is that they have variable oxidation states. This is because the five inner d orbitals are at a similar energy level to the outer s orbital. In the transition elements, d electrons as well as s electrons are involved in bonding. Ionic bonds form when 4s and then 3d electrons are lost to produce positively charged ions. Covalent bonds are formed as unpaired electrons pair up with those on other atoms.
The table below shows the main oxidation states (bold being the most important) and the electronic structures of the elements titanium to copper.
|
Element |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
|
|
|
|
|
|
|
|
|
+1 |
|
|
+2 |
+2 |
+2 |
+2 |
+2 |
+2 |
+2 |
+2 |
|
Oxidation |
+3 |
+3 |
+3 |
+3 |
+3 |
+3 |
+3 |
|
|
states |
+4 |
+4 |
|
+4 |
|
|
|
|
|
|
|
+5 |
|
|
|
|
|
|
|
|
|
|
+6 |
+6 |
+6 |
|
|
|
|
|
|
|
|
+7 |
|
|
|
|
Complex Ions
Transition metals form
compounds containing complex ions, also known as coordination compounds. Such
complex ions are formed by the coordination of lone pairs of electrons from a
donor (called a ligand) to an atom
or cation (called an acceptor) which
has empty orbitals to accommodate them. A cation may form a complex with a
neutral molecule, e.g.
[Cu(NH3)4(H2O)2]2+
or with an oppositely
charged ion, e.g.
[CuCl4]2-
An atom may form a
complex, e.g., Ni(CO)4. The charge remaining on the central atom or
ion when the ligands are removed together with their lone pairs is the oxidation number of the metal in the
complex. The coordination number is
the number of atoms forming coordinate bonds with the central atom or ion: 2, 4
and 6 are common. One definition of a complex ion involves these terms: a
complex ion is an ion in which the coordination number exceeds the oxidation
number of the central atom.
Ligands
must possess one or more unshared pairs of electrons. A ligand which can only
form one bond to a central atom or ion is called a monodentate (literally, 'one tooth') ligand, e.g., NH3 ,
CN-. A polydentate ('many
teeth') ligand can form more than one bond. Examples are the ethanedioate ion,
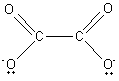
and ethane-1,2-diamine
(known as ‘en’ for short),
![]()
in which the lone pairs on the two nitrogen atoms can form coordinate bonds. The two coordinate bonds formed by each ligand are thought to resemble the claws of a crab (Greek: chele), and such compounds are named chelate compounds or chelates. In the polydentate ligand, 1,2-bis[bis(carboxymethyl)amino]ethane
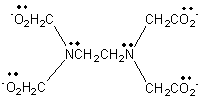
which is called edta
(short for its old name, ethylenediaminetetraacetate), there are unshared
electron pairs on four oxygen atoms and on the two nitrogen atoms. This ligand
forms six coordinate bonds, and its complex ions are very stable.
Nomenclature:
how to name transition metal compounds
In the formula of a complex ion, the symbol for the central atom appears first and is followed by the anionic ligands and then by neutral ligands, e.g.
[CoCl2(NH3)4]2+
The
formula of the complex ion may be enclosed in square brackets.
The name of the complex gives the name and oxidation state of the central metal cation, e.g., cobalt(III), preceded by the name and number of ligands attached to it, e.g.,
[Co(NH3)6]3+ is the hexaamminecobalt(III)
ion
The prefixes di tri
tetra penta hexa
are used to show the number of ligands. If several ligands are present, they are listed in alphabetical order, and the prefixes, di, tri, etc., are not allowed to alter this order, e.g.
[CrCl2(H2O)4]+ is the tetraaquadichlorochromium(III)
ion
If the complex is an anion, the suffix -ate follows
the name of the metal, e.g., zincate and chromate. If the metal has a Latin
name, then in the complex anion the Latin name of the metal is used, followed
by the suffix -ate, e.g.
[Fe(CN)6]4- is
the hexacyanoferrate(II) ion
Examples are given in the table below:
|
Ligand |
Type of complex |
Example |
Name of Complex |
|
Water |
Aqua- |
[Cr(H2O)6]3+ |
Hexaaquachromium(III) ion |
|
Ammonia |
Ammine- |
[Ag(NH3)2]+ |
Diamminesilver(I) ion |
|
Hydroxide ion |
Hydroxo- |
[Zn(OH)4]2- |
Tetrahydroxozincate(lI) ion |
|
Chloride ion |
Chloro- |
[CuCl4]2- |
Tetrachlorocuprate(lI) ion |
|
Cyanide ion |
Cyano- |
[Fe(CN)6]3- |
Hexacyanoferrate(III) ion |
|
Nitrite ion |
Nitro- |
[CO(NO2)6]3- |
Hexanitrocobaltate(III) ion |
|
Carbon monoxide |
Carbonyl- |
Ni(CO)4 |
Tetracarbonylnickel( 0) |
|
Ethane-l,2-diamine |
Ethane-I,2-diamine- |
[Cr(en)3]3+ |
Tris(ethane-l,2-diamine)chromium(III) ion |
|
edta |
edta- |
[Zn(edta)]2- |
edtazincate(lI) ion |
|
Cl-, NH3 |
Mixed |
[CoCl2(NH3)4]+ |
Tetraamminedichlorocobalt(III) ion |
|
|
Mixed |
[Fe(OH)2(H2O)4]+ |
Tetraaquadihydroxoiron(III) ion |
The
shapes of complex ions
The spatial arrangement of bonds from the ligands to the central atom or ion depends on the identity of the atomic orbitals used by the central atom or ion and on the coordination number of the transition element. The most common shapes are octahedral, tetrahedral, square planar (nickel and platinum) and linear (silver) complex ions.
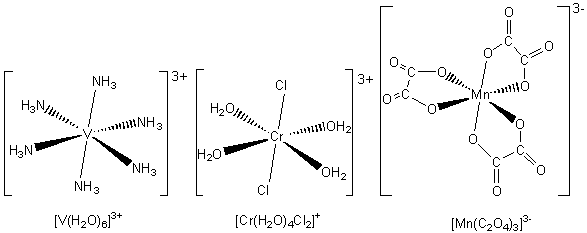
Octahedral complexes: six-fold coordination
An octahedral complex results when there are six bonds to the central metal ion. This can happen if there are six monodentate ligands, three bidentate ligands, or other combinations that give the six bonds. Three examples of octahedral complexes are shown below.
Tetrahedral complexes: four-fold
coordination
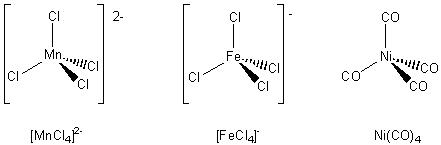
A tetrahedral complex has
four bonds from the ligands to the central metal ion. The ligands lie at the
four corners of a tetrahedron. Three examples are given below.
Square planar complexes:
four-fold coordination
Platinum and nickel are unusual in that they form square planar complexes. Two examples are given below. A square planar complex also has four bonds from the ligands to the central metal ion but this time the metal ion and the four ligands all lie in the same plane. A square planar compound, cisplatin, is an anti-cancer drug that has a remarkable success rate in curing testicular cancer. Cisplatin, diamminedichloroplatinum(II), is a complex of platinum(II), Pt(NH3)2Cl2, having chloride ions and ammonia molecules as ligands.
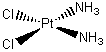
Linear complexes: two-fold coordination
Silver ions are unusual in having a preference for forming linear complexes. In these, the silver ion is bonded to two ligands only and the bond angle is 180°. Examples of these complexes and their uses are:
[Ag(NH3)2] +
This is formed when silver chloride or silver oxide dissolves in aqueous ammonia to form the colourless solution known as Tollen's reagent. This reagent is used to distinguish aldehydes from ketones; aldehydes give a silver mirror on the side of the test tube:
![]()
[Ag(S2O3)2] 3-
This ion is formed when
silver salts are dissolved in sodium thiosulphate ("hypo") solution.
This reaction is important in photographic fixing. The silver bromide which has
not been exposed to light is dissolved away from the film leaving the black
image of silver as the negative:
![]()
[Ag(CN)2] -
This complex ion is formed when silver salts are dissolved in solutions of sodium or potassium cyanides. This solution is used as the electrolyte in silver plating. The deposition potential for silver is greatly raised over that for deposition from aqueous silver solutions so that silver can only be displaced when a sufficiently high potential is applied. Under these circumstances, the silver deposits in a compact and adherent form.
Predicting the shape of transition metal complexes
Most
transition elements form both octahedral and tetrahedral complex ions. But when
is each formed? The important factors are the size and charge of the ligand.
Lots of small, neutral ligands (e.g. NH3, H2O) can crowd
around a central metal ion, so these ligands tend to form octahedral complexes.
On the other hand, fewer large or negatively charged ligands (e.g.
For example, when a pink, aqueous solution of cobalt(II) sulphate, containing [Co(H2O)6]2+, is treated with an excess of concentrated hydrochloric acid, a deep blue solution is formed, containing [CoCl4]2-.
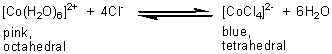
If the blue solution is diluted with water, the pink solution is reformed. Likewise, when concentrated hydrochloric acid is added to an aqueous solution of copper(ll) sulphate which contains the blue complex ion, [Cu(H2O)6]2+, an olive-green solution containing [CuCl4]2- is formed:
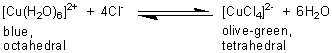
If the solution is diluted, then the increased concentration of water forces the equilibrium back to the hexaaqua ion and the solution returns to a blue colour.
Note: if we decrease the size of the ligand to that of F-, then octahedral complexes become common e.g. AlF63- (compare with AlCl4-); if, however, we increase the size of the ligand to that of I-, we find no octahedral complexes with any of the cations of first three periods.
Isomerism in complex
ions
An ion or molecule has isomers if it can exist in two or more different forms with the same formula. Isomers frequently occur in organic compounds, but one of the fascinating things about transition metal chemistry is that many complex ions have isomers. The study of isomerism in complexes was one of the main interests of the German chemist Alfred Werner. We owe much of our understanding of transition metal complexes to him. In his book New Ideas on Inorganic Chemistry published in 1911 he discussed the main types of isomerism. We shall follow some of his examples, although we shall change the names that he used to their modern versions.
Ionisation isomerism
This
is the first type of isomerism we shall look at. Werner says:
A peculiar type of isomerism. . . is that in
which compounds of the same composition yield different ions in solution.
His example was two compounds with the formula Co(NH3)5BrSO4.
Werner noted that in solution:
The one is a reddish-violet colour, and
gives in a freshly prepared solution no reaction for bromide ions, but does for
the sulphate ion. On the other hand, the solution of the second salt gives the
reactions of bromide ion, but not of the sulphate ion.
Well, so much for the facts. How can we explain them? The difference is due to the way the bromide and sulphate are bonded in the compounds. In both of them the five ammonia molecules are ligands. This leaves space for one other ligand. In one of the complexes the bromide is the ligand, in the other it is the sulphate ion. The two possibilities are shown in below. In the first one the bromide is strongly bonded to the cobalt ion. We say that the ammonia molecules and the bromide ion are in the coordination sphere. The sulphate ion is held in the crystal with its two negative charges just balancing the two positive charges on the complex.
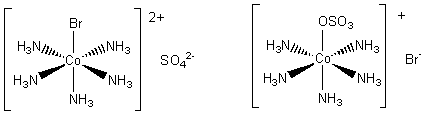
When the solid is put in water the sulphate ion floats away, but the bromide is left attached to the cobalt. Therefore, if we test for the presence of sulphate ions we find a positive result, but a test for free bromide ions is negative. With the second variety of the salt, the opposite is true. The sulphate ion is bonded to the cobalt, and free bromide ions can be found in the solution. (The sulphate ion is now in the coordination sphere with the ammonia molecules.)
Hydrate isomerism
This is the second type of
isomerism that we need to know about. Werner comments:
The most beautiful example of hydrate
isomerism is that furnished by chromium 'chloride. Three different hydrates of
the formula CrCI3 + 6H2O are known. One is greenish-blue,
which dissolves giving a blue-violet solution, and two are green, which
dissolve yielding green solutions. ..
The
difference between the three hydrates is shown by writing their formulae as:
[Cr(H2O)6]3+ (Cl-)3 blue-violet in solution
[Cr(H2O)5Cl]2+ (Cl-)2 green in solution
[Cr(H2O)4Cl2]+ Cl- darker green in solution
You
should understand why it is that one mole of each of them give three, two and
one moles of chloride ions in solution respectively. When they are crystallised
from solution the second takes up one mole and the third takes up two moles of
water of crystallisation. This gives them all the overall formula CrCI3•6H2O.
Geometrical isomerism
This
is the third type of isomerism. Werner points out that a complex ion like
[Co(NH3)5CI]2+ has no isomers. However, two
isomers of [Co(NH3)4CI2]+ can be
identified:
Up to the present, and in spite of much careful work, it has been found
impossible to obtain three isomers. These facts are best explained by assuming
that the groups are placed around the central atom at the corners of a regular
octahedron.
The ion [Co(NH3)5CI]2+ is an octahedral complex. Because of the symmetry of the octahedron all the diagrams below represent the same complex.
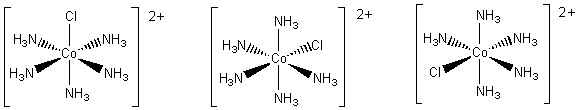
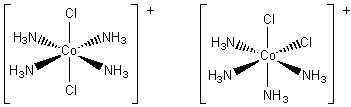
If an ammonia molecule is replaced by a chloride ion we have [Co(NH3)4CI2]+. This time there are two possible structures. In one of them the four ammonia molecules and the cobalt ion lie in a plane. The chloride ions lie above and below the plane, in the axial positions. In the second, a plane containing the cobalt ion also contains three ammonia molecules and one of the chloride ions. This leaves the remaining ammonia molecule and chloride ion in the axial positions. The two arrangements are different, as illustrated in below. They are geometrical isomers.
Square planar complexes
can also have geometrical isomers. The diagram below shows two isomers that
have the formula Ni(NH3)2Cl2. When the two
chlorides (or ammonia molecules) are on the same side of the square, they are
said to make the cis isomer (left)
When they are opposite one another, we have the trans isomer (right).
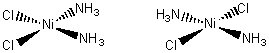
Optical isomerism
This is the final type of isomerism we shall meet here. An optical isomer rotates the plane of polarised light. If it rotates the plane to the left, it is called an I form (I for laevo, left). If it rotates it to the right, it is called a d form (d for dextro, right). A test to discover if a molecule or ion is optically active is to see if it can be superimposed on its mirror image. Examples that occur in transition metal chemistry are octahedral complexes involving a bidentate ligand such as ethane-1,2-diamine. The three ligand molecules can attach themselves in two ways, shown below, which resemble aeroplane propellers.
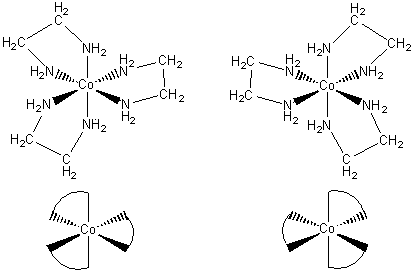
Two
isomers of [Co(en)3]3+, which are mirror images of each
other, are known. They are optical isomers. The top two diagrams show the
arrangements of the atoms; the bottom two use curved lines to represent the
ligands.
What happens to the d
orbitals in a complex ion?
There
are two main theories of bonding that are useful for explaining the behaviour
of transition metal complexes. The first is crystal field theory, the
second is ligand field theory.
In crystal field theory we imagine that the ligands act as centres of electric charge. Then we try to work out what effect the field of the ligands has on the electrons in the d orbitals. This was the first theory invented to explain the properties of complexes. Given that it ignores the finer, and even the important, points of bonding, it is remarkably successful. This is what it has to say about he effects on the d orbitals.
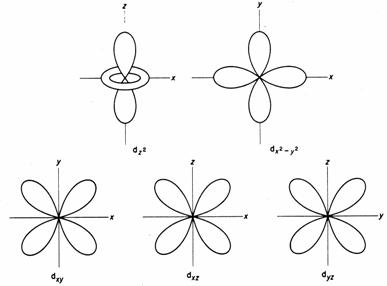
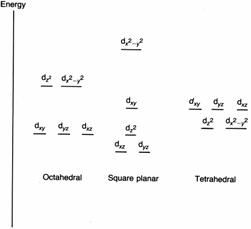
First, if you bring negative charges near to electrons, the energies of the electrons rise. This has the effect of increasing the energies of all the d orbitals. Now let us look at the effects on the individual orbitals. The diagram below shows the shapes of the d orbitals. Let us make a diagram of an octahedral complex and show the dz2 and dx2-y2 orbitals as well. The lobes of the two orbitals point directly at the ligands. Electrons in these orbitals will suffer a significant amount of repulsion. Now look at the next diagram, which shows the arrangements of the dxy, dxz and dyz orbitals.
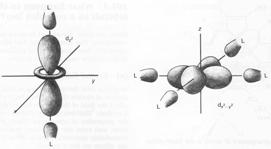
Ligands at the ends of the x, y, and z axes have electron density that
will interact strongly with electrons in dz2 and dx2-y2
orbitals
These
have their lobes pointing between the ligands. As a result, electrons in these
orbitals feel less repulsion than those in the other two orbitals. Thus crystal field theory says that
the five d orbitals should split into two groups, two of which have a higher
energy than the other three.
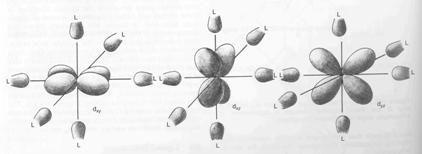
Ligands
at the ends of the x, y, and z axes have weaker interactions with dxy,
dxz and dyz orbitals than with dz2 and dx2-y2
orbitals
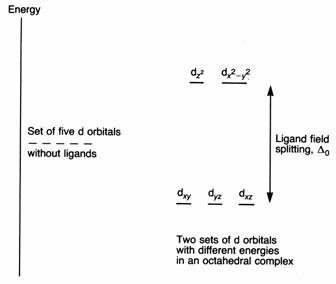
We say that the d orbitals are split. Owing to the splitting being caused by the electric field of the ligands, it is called the ligand field splitting (see diagram). The extent of the splitting is given the symbol Δo. The two orbitals with the higher energy are called the eg set; the three with the lower energy are the t2g set.
Ligand field theory
In ligand field theory we take a more realistic view of the bonding in complexes. We use molecular orbital theory to decide how the orbitals on the metal ion and the ligands interact. As you might expect, this is rather complicated. However, the nice thing is that ligand field theory comes up with the same scheme of splitting as crystal field theory, so we do not need to discuss it further. The two theories also agree on the splitting patterns for d orbitals in tetrahedral and square planar complexes which are quite different to the octahedral case, see below.
Colour
of Transition Metal Complexes
An introduction to colour
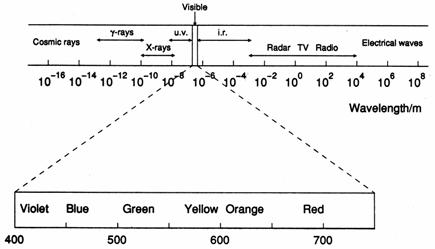
The electromagnetic spectrum comprises the total range of physically available frequencies of radiation. High-energy radiations, such as γ-rays and X-rays, are of very short wavelengths whereas low-energy radiations, such as radio waves and electrical waves, are of very long wavelengths. The visible region is situated between the ultra-violet and the infra-red regions and occupies only a very narrow portion of the total spectrum, extending from about 400nm to 750nm in wavelength.
Regions
of the electromagnetic spectrum
Light has dual characteristics (wave-particle duality). Maxwell first suggested that light was electromagnetic in character, consisting of mutually perpendicular fields whose amplitudes varied in a wave-like manner. The wave front travels with a speed of approximately 3.00 x 108 ms-1. Light can be characterised either by its wavelength (λ), the distance required for one complete oscillation of the wave, or by its frequency (ν), the number of oscillations occurring in unit time (Hz = s-1). The speed of the wave front is given by the product of these two quantities:
c = λ ν
Wavelength
and frequency are, of course, inversely proportional. The wave theory is most
successful in accounting for the propagation of light.
The particle theory of light was put forward in 1905 by Planck and Einstein who regarded light as a stream of discrete particles of energy (photons), travelling at the same speed as that of the wave front in the wave theory. In the particle (quantum) theory, monochromatic radiation is characterised by the energy of each photon. The photon energy (E) is related to the frequency of the radiation by the Planck-Einstein equation:
E = hν
The proportionality constant (h), the Planck constant, has a value of 6.626 x 10-34 Js. The energy associated with one mole of photons is obtained by multiplying hν by the Avogadro constant (L), which has a value of 6.022 x 1023 mol-1. The quantum approach best explains how light interacts with matter.
Electronic excitation (E1-
E0 = hν)
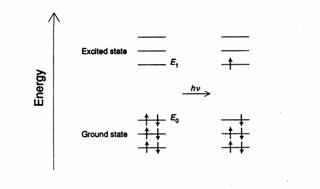
Because the
electrons in atoms and molecules exist in discrete (specific) energy levels, absorption
of radiation from the ultra-violet and visible regions of the electromagnetic
spectrum leads to the excitation (promotion) of valency electrons in the
low-energy ground state to the higher energy excited state. The smallest energy
change is that associated with the excitation of an electron from the highest
energy level in the ground state to the lowest energy level in the excited state.
The same energy difference (Δ) applies in the emission of radiation
where an electron in the lowest excited level falls to the highest ground
level.
Electronic absorption spectra are usually measured in solution and generally appear as broad bands rather than as sharp peaks. This is because molecular vibrations and rotations very slightly alter the energy of each molecule in a sample.
The
absorption spectrum of Malachite Green in ethanoic acid
Predicting the colour of
substances: the colour circle
All coloured substances absorb radiation selectively from the visible region in a way which is characteristic of the molecular species concerned. It should be realised that absorption in the ultra-violet is characteristic of colourless organic substances. In both cases, electronic transitions are involved. Coloured species usually absorb in the ultraviolet as well as in the visible region.
|
Wavelength/nm |
Colour
absorbed |
Visible colour |
|
400 - 435 |
Violet |
Yellow-green |
|
435 - 480 |
Blue |
Yellow |
|
480 - 490 |
Green-blue |
|
|
490 - 500 |
Blue-green |
Red |
|
500 - 560 |
Green |
Purple |
|
560 - 580 |
Yellow-green |
Violet |
|
580 - 595 |
Yellow |
Blue |
|
595 - 605 |
|
Green-blue |
|
605 - 750 |
Red |
Blue-green |
It is possible to trace a relationship between the colour of light absorbed and the related visible colour when the compound has a narrow single absorption band within the wavelengths listed. When light of a given colour is absorbed, the complementary colour is transmitted or reflected. Thus, for example, a dye or a pigment absorbing light within the yellow region will appear blue to the eye in daylight. The nine regions of the visible spectrum can be depicted in the form of a colour wheel or colour circle. Just as white light can be split into its coloured components by means of a prism, the spectrum colours can be recombined to produce white. White light is also produced by mixing two complementary colours from any pair of opposite sectors in the colour circle. Mixing radiations in this way is called additive mixing. Any colour can be produced by a suitable additive mixture of red, green, and blue light (primary colours).
The colour purple is not duplicated by any single wavelength of light. This so-called non-spectral colour can be produced by mixing red and violet monochromatic radiations.
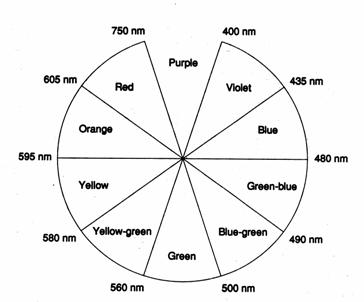
The
colour circle of complementary colours
Most
coloured substances of interest owe their appearance to a subtractive process.
As mentioned above, absorption (removal) of yellow light leads to a blue
appearance; the converse is true. The same effect can be achieved by passing
white light through appropriate colour filters, as in a colorimeter. The
production of green by a subtractive process is of special interest because
purple does not exist as monochromatic radiation. However, removal of both red
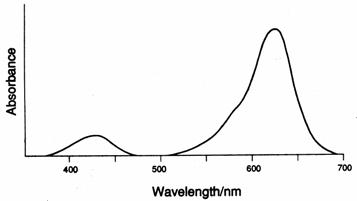
and violet light gives rise to green. The dye Malachite Green is a good example
(see the absorption spectrum, above). Thus, absorption of violet (427.5nm) and
red (621nm) light results in the appearance of green. Simultaneous absorption
in two regions of the visible spectrum also occurs with chlorophyll, the most
abundant green pigment, which absorbs at wavelengths close to 430nm and 660nm.
Bright, pure colours are produced by dyes having narrow absorption bands.
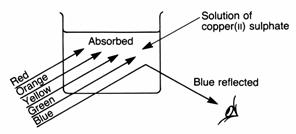
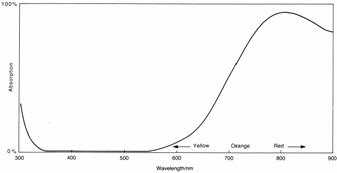
Some substances absorb all of the colours of the visible spectrum except for a narrow range of wavelengths which are reflected back to the observer. For example, if copper(II) sulphate is placed in sunlight, i.e. white light, the solution looks a royal blue. If, however, it is placed in a photographic darkroom that has a red safety light, the solution looks black. The reason for this is that copper(II) sulphate reflects blue light very efficiently; but it absorbs light of most other colours, especially light in the red area of the visible spectrum (see absorption spectrum, below). This means that in the dark room, where there is only red light shining on the solution, the photons of red light are absorbed by the copper(II) sulphate. There are no photons of blue light to be reflected by the solution, so no light reaches our eyes and the solution looks black.
Substances with broad absorption bands which cover a significant proportion of the visible region tend to give dark or dull hues, such as maroon or brown. Black results from absorption across the whole of the visible spectrum since all the spectral colours are removed.
Why are transition metal compounds coloured?
Transition metal complexes are coloured because visible light has just about the right energy to excite an electron in the lower set of d orbitals into the higher set (a d→ d transition). All substances in which the central metal ion has an incomplete set of d electrons (3d1 to 3d9 for the first transition series) will be coloured, whether in an octahedral or tetrahedral ligand field. For example, the octahedral complex hexaaquanickel(II), [Ni(H2O)6]2+, is responsible for the green colour of many nickel(II) salts in water (see diagram below). The complex appears green because nickel(II) (3d8) absorbs light in the red, orange, yellow and blue regions of the visible spectrum, allowing only the green light to be reflected back to the observer. In this case, the splitting of the d orbitals corresponds to the energy of blue light (400nm), so this wavelength is absorbed when an electron is promoted from the t2g set of orbitals to the eg set. The other wavelengths are absorbed by electronic transitions including transfers of electrons between the ligand and the metal ion and by the vibrations of the bonds in the complex.
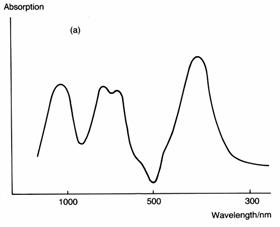
The
spectrum (a) of [Ni(H2O)6]2+ shows a number of
strong absorptions. However, the complex absorbs only weakly around 500nm,
which is the green and blue-green region of the visible spectrum. The ligand
field splitting (b) in [Ni(H2O)6]2+ matches the
frequency of light in the blue region of the spectrum
Obviously, compounds in which the metal has a full set of d electrons (e.g. 3d10) or, indeed, no d electrons (3d0) cannot give rise to d→ d electron transition. However, colour can arise in these species by charge transfer where an electron between the ligand and the metal, resulting in an intense absorption. For example, copper(I) oxide, Cu2O, in which copper(I) has a full 3d10 subshell is red due to charge transfer.
The colours of transition metals are directly related to the energies of d→ d transitions, governed by the ligand field strength, Δ. This is affected by:
- the number of ligands
- the shape of the complex
- the oxidation state of the metal
- the nature of the ligand
We have already seen how the number of ligands and the shape of the complex affects the colour of copper(II) and cobalt(II) complexes. For example,
The ligand
field strength in a tetrahedral field, Δtet, is less than that
in an octahedral field, Δo, so the complex absorbs light of
lower energy when an electron undergoes a d→ d transition. The
hexaaquacobalt(II) ion absorbs light in the blue-green (490nm) part of the
visible spectrum and therefore appears as the complementary colour, pink. The
tetrachlorocobaltate(II) ion absorbs light in the lower-energy, yellow region (590nm)
of the spectrum and appears as the complementary colour, blue.
An increase in oxidation state of the central metal tends to increase the ligand field strength, Δo. For example, the blue hexaaquachromium(II) is readily oxidised to the ruby-red hexaaquachromium(III) in air:
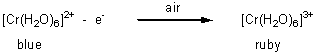
the hexaaquachromium(II) ion absorbs light in the yellow part of the visible spectrum (590nm) and so it appears as the complementary colour, blue. However, the hexaaquachromium(III) ion absorbs blue-green light in a higher energy part of the spectrum (490nm) so it appears as the complementary colour, red
In general, the ligand field strength, Δo, for d→ d transitions in octahedral complexes increases in the order:
Mn2+
< Ni2+ < Co2+ < Fe2+ < V2+
< Fe3+ < Cr3+ < V3+ Co3+
As might be expected, the higher the charge on the metal ion, the stronger the interactions with ligands in the crystal field. This is the spectrochemical series for metal ions.
Ligand substitution can have a very dramatic effect on the colour of a compound. Treating the ruby-red hexaaquachromium(III) ion with concentrated ammonia solutions leads to the formation of the yellow hexaamminechromium(III) ion:
![]()
The hexaaquachromium(III) ion absorbs light in the blue-green region (490nm) -because of a d→ d transition and so it appears red. The hexaamminechromium(III) ion has a stronger ligand field, so it absorbs higher energy blue light (460nm) and appears as the complementary colour, yellow.
A spectrochemical series for ligands, in order of increasing ligand field strength is:
I-
< Br- < Cl- < S2- < F-
<
Why are compounds of transition elements often paramagnetic?
Most people know
that iron is magnetic. In fact, iron shows a very special type of magnetism
called ’ferromagnetism’. The two metals following it in the first transition
series, cobalt and nickel, also show this type of magnetism. Ferromagnetism is
a variety of magnetism known as paramagnetism. A paramagnetic substance
put close to a magnetic field will be attracted into the field. Many transition
metal compounds are paramagnetic.
It is the magnetic field associated with the spin of an electron that can give rise to paramagnetism. However, only compounds that have unpaired electrons are paramagnetic. When two electrons occupy the same orbital, they have their spins paired, and we can think of the magnetic fields cancelling out. To sum up: transition metals are paramagnetic when they have one or more unpaired electrons.
If we measure the amount of their paramagnetism we can work out how many unpaired electrons are present. This can be done using a Gouy balance (see diagram). An empty glass tube is suspended between the jaws of an electromagnet. Then the tube is filled with crystals of the chemical being investigated. The magnet is switched on and, if the substance is paramagnetic, the tube is pulled down into the field. Masses are placed, on the balance pan until the balance is zeroed again. The mass used is a measure of the amount of paramagnetism, and therefore the number of unpaired electrons. If we are to understand how these unpaired electrons come about, we need to look carefully at what happens to the d orbitals when a complex ion is made.
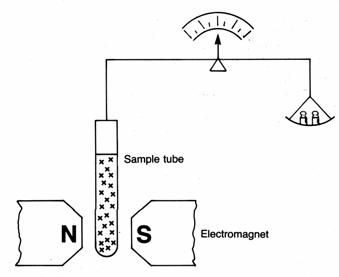
The principle of the Gouy balance: the sample is balanced with the
electromagnet off, and again with it on. The difference between the two
weighings is a measure of the number of unpaired electrons in the sample.
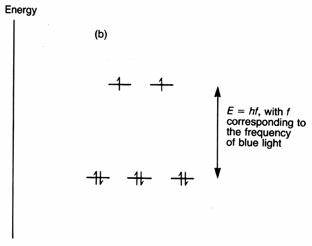
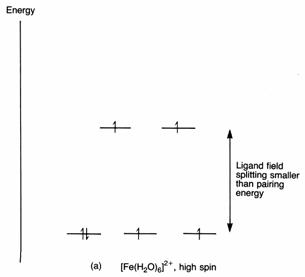
The two complexes of iron(II), hexaaquaferrate(II), [Fe(H2O)6]2+ and hexacyanoferrate(II), [Fe(CN)6]4-, are both octahedral. The first has paramagnetism corresponding to four unpaired electrons. The second is not paramagnetic; it has no unpaired electrons. Now, the electron structure of iron is [Ar]3d64s2. When it loses two electrons, it is left with the argon core and six 3d electrons. We have to arrange these six electrons on the energy level diagram for an octahedral complex. We know that electrons will go into separate orbitals whenever possible so that the repulsion among them is minimised. If we follow this pattern we shall have one electron in each of the five orbitals, and one left over. This will go into the orbital of lowest energy. Making sure that we stick to the Pauli principle, the two electrons in the. same orbital will have their spins paired. This is the arrangement shown below. You can see that there are four unpaired electrons, just as found from experiment.
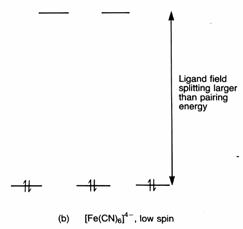
Cyanide ions cause a much larger splitting than do water molecules. One
result is that cyanide complexes are often ‘low spin’.
In the case of [Fe(CN)6]4-
there are no unpaired electrons. The only way of achieving this, and keeping
the energy of the electrons to a minimum, is to put them as three pairs in the
t2g set. This is the arrangement shown below. The reason why they
take up this arrangement is that cyanide ions have a larger effect on the
splitting of the d orbitals than do water molecules. The ligand field splitting
is larger in [Fe(CN)6]4- than [Fe(H2O)6]2+.
When the gap Δo becomes large, it takes more energy to put an
electron into one of the eg set than it does to pair electrons in
the t2g orbitals. For fairly obvious reasons, [Fe(H2O)6]2+
is called a high spin complex, and [Fe(CN)6]4- a low
spin complex.
Finding the formula of a complex by colorimetry
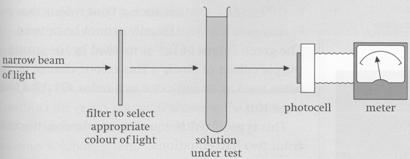
The colour of a transition metal ion can change as the ligand is changed, as we have seen above. We can use this change in colour to find the ratio of ligand to metal in a complex, using a colorimeter. In a colorimeter a narrow beam of light passes through the solution under test towards a sensitive photocell (see diagram). The wavelength of this light can be selected by using an appropriate filter. The current generated in the photocell is proportional to the amount of light transmitted by the solution, which depends on the colour of the solution. The colorimeter is usually calibrated to show the fraction of light absorbed by the solution - the most intensely coloured solution absorbs the most light, and the faintest coloured solution absorbs the least light.
Let's use an example to see how this method works. A solution of Fe3+ ions, such as aqueous FeCl3, is yellow. The complex ion is actually [Fe(H2O)6]3+. When aqueous sodium thiocyanate, NaSCN, is added the complex turns blood-red as the thiocyanate ligands (SCN-) displace the water ligands. How do we work out the formula of the Fe3+/ SCN- complex? Ten tubes are prepared which contain different amounts of Fe3+ and SCN-. The tube with the most intense colour, as read by the colorimeter, contains the maximum amount of ligand molecules to metal ions. The table contains details of one method you can follow. The principles of the method are always the same, although the volumes and concentrations of the metal ion and ligand solutions can change. What is extremely important is to use the same volume of solution in each tube.
|
Tube |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
vol. in cm3
of 0.1 moldm-3 Fe3+(aq) |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
|
vol. in cm3
of 0.1 moldm-3 SCN-(aq) |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
|
vol. in cm3
of H2O added to make final volume 40cm3 |
28 |
26 |
24 |
22 |
20 |
18 |
16 |
14 |
12 |
10 |
The Fe3+ and SCN- solutions are measured as accurately as possible into the tubes. The final solution from each tube in turn is then placed in a test-tube or cuvette and placed in the colorimeter. The complex is red so it absorbs in the blue region of the spectrum, so a blue filter is used, showing that the complementary colour to the complex is used in the filter. The absorbance reading is taken. The results are shown below.
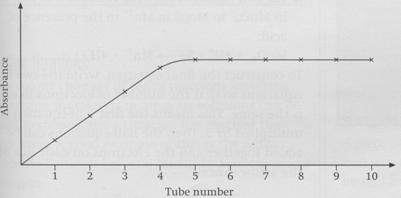
As you can see, the absorbance readings taken on the
colorimeter go up as the red colour gets more intense, and finally the
readings level out. The point where the horizontal line is reached shows the
number of moles of Fe3+ and SCN- which form the complex.
In this case it is at tube 5. In this experiment, this solution contains 10 cm3
of 0.1 moldm-3 Fe3+ and 10 cm3 of 0.1 moldm-3
SCN-. From these values you can work out the amount in moles of each
species: 0.001 mol Fe3+ and 0.001 mol SCN-. So the ratio
is 1 Fe3+ : 1 SCN- and the formula of the complex is
therefore [Fe(H2O)5(SCN)]2+.
Transition
elements in paints
Transition
metal complexes are often used in paints to make many coloured pigments. In
fact, for brilliance and clarity of colour, transition metal complexes with
organic ligands cannot be bettered. Examples of such pigments include Monastral
Blue, in which copper(II) ions (d9) are complexed by phthalocyanine
ligands. If hydrogen atoms in phthalocyanine are replaced by bromine or
chlorine atoms, the colour of the pigment can be changed to other blue shades
or various shades of green.
Titanium(IV) oxide, TiO2,
includes a titanium(IV) ion with the electronic configuration [Ar]3d04s0. Obviously,
no d electrons are present and so d→ d transitions are impossible.
Titanium(IV) oxide therefore reflects all incident light and is white. It is
used as the pigment for most white paints.
How do the transition elements act as catalysts?
Transition elements can
act as catalysts for two reasons. First, because they have different oxidation
states, they can take part in electron transfer reactions. One example that
demonstrates this is the effect of adding iron(II) or iron(III) ions to a mixture
of peroxodisulphate(VI) ions, S2O82-, and iodide
ions, I-. Normally the reaction:
S2O82-(aq) + 2I-(aq) → 2SO42-(aq) + I2(s) (A)
takes place at a convenient rate, which we can follow by one of the usual methods. If a little iron(III) sulphate solution is added, the rate of production of iodine increases markedly. This is because the Fe3+ ions react with the iodide ions:
2Fe3+(aq) + 2I-(aq) →
2Fe2+(aq) + I2(s) (B)
If this were all that happened, we would not be
dealing with true catalysis: the Fe3+ ions appear to be used up in
the reaction. However, the Fe2+ ions are oxidised back to Fe3+
by S2O82- ions:
2Fe2+(aq) + S2O82-(aq) →
2Fe3+(aq) + 2SO42-(aq) (C)
If you combine equations (B) and (C) you will obtain the original equation, (A). The Fe3+ ions have taken part in the reaction, but they are regenerated.
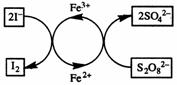
The key point is that by giving a different path by
which electron transfer can take place, the ions have provided a route with lower
activation energy. Reaction (A) takes place between ions of with the same charge
which repel each other. Reactions (B) and (C) are between ions of opposite
charge which attract each other and the reactions are therefore faster.
Transition metals and
their ions can also act as catalysts by providing sites at which reactions can
take place. They can bond to a wide range of ions and molecules, e.g. those
with lone pairs or π electrons. Also they show the ability to make
different numbers of bonds, often four or six, but sometimes two, three and
five as well. They can show catalytic behaviour when dissolved in solution or
as solids, i.e. as homogeneous or heterogeneous catalysts.
An important example of a catalytic reaction in solution is
the Ziegler-Natta polymerisation of alkenes (see below). A typical
Ziegler-Natta catalyst is a mixture of an organic aluminium compound, e.g. Al(C2H5)3,
and titanium(III) chloride. One of the ethyl groups from the aluminium compound
bonds to the titanium ion; but being a transition metal ion, it has further sites
available for bonding. The π cloud of an alkene can fill one of these
sites, with the result that the alkene and ethyl group are held very close to
one another. For reasons that we do not have to consider, the alkene inserts
itself between the ethyl group and the titanium ion. Now there is room for a
further alkene to bond to the titanium ion, which in turn swaps its position.
In this way a hydrocarbon chain that originally had only two carbon atoms in it
now has six of them joined together. Provided the supply of alkene molecules
is kept up, the chain will grow to great length. In other words, a polymer is
made. The discovery of this type of reaction by Ziegler and Natta has
revolutionised the manufacture of polymers, partly because the method can give
polymers of consistent quality.
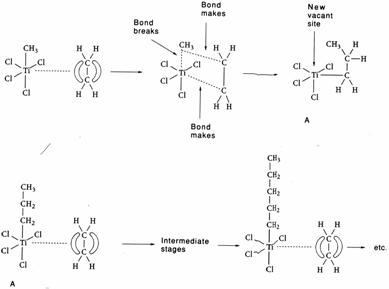
These diagrams show the type of
change that takes place with Ziegler-Natta catalysts. The first step is an
alkene bonding to a titanium complex through its π cloud of electrons.
Then there is a rearrangement of the atoms, which leaves a complex, A, that can
repeat the process. The result is growth of the polymer chain.
The chemistry of selected transition
elements
Vanadium
The chemistry of vanadium provides us with one of the most striking illustrations of variable oxidation states. The highest oxidation states occur when the element combines with the highly electronegative element oxygen. Vanadium shows its oxidation number +5 in the vanadate(V) ion. When zinc reduces ammonium vanadate(V) in concentrated hydrochloric acid, the solution changes from yellow to blue-green, and eventually violet as the oxidation number changes from +5 through to +2:
VO22+(aq) → VO2+(aq) → V3+(aq) → V2+(aq)
V(V) V(IV) V(III) V(II)
yellow blue blue-green violet
A stereotypical transition element, vanadium also displays catalytic properties. It is used to catalyse the oxidation of sulphur(IV) oxide in the Contact Process.
Vanadium and the manufacture of sulphuric acid (the Contact Process)
Sulphuric
acid is manufactured by oxidising sulphur dioxide to sulphur trioxide. This
intermediate product is then converted into sulphuric acid.
(i)
(a) burning sulphur in air:
S(l)
+ O2(g) ![]() SO2(g) DHø=
-298kJmol-1
SO2(g) DHø=
-298kJmol-1
or
(b) burning H2S from crude oil in air.
(ii)
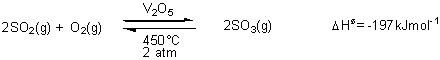
According to Le
Chatelier’s Principle, the exothermic reaction will be favoured by low
temperatures and high pressures. At pressures above 2atm, however, the sulphur
dioxide liquefies; at low temperatures, the rate of attainment of equilibrium
is slow because the forward and reverse reactions have a large activation
energy. The use of a catalyst (V2O5) allows compromise conditions (450°C and
2atm) to be used whilst obtaining an economically viable yield.
(iii) Once sulphur trioxide, SO3, has been prepared, it is reacted with water produce sulphuric acid:
SO3(g) + H2O(l) ![]() H2SO4(l)
H2SO4(l)
In fact, this reaction is not used much in
industry in
SO3(g)
+
H2SO4(l) ![]() H2S2O7(l)
H2S2O7(l)
The oleum is then converted into more sulphuric acid by adding water:
H2S2O7(l) + H2O(l) ![]() 2H2SO4(aq)
2H2SO4(aq)
The contact process is used to make about
156 million tonnes each year of sulphuric acid (2.3 million tonnes in the
Vanadium is able to catalyse the reaction because it has variable oxidation states. Firstly, vanadium(V) oxide reacts with sulphur dioxide to form sulphur trioxide and sulphur trioxide:
V2O5 + SO2 → V2O4 + SO3
The vanadium(V) oxide is then regenerated (a requirement for catalysis) by reaction with oxygen:
2V2O4 + O2 → 2V2O5
These two steps each have a much smaller activation energy than the direct, uncatalysed oxidation of sulphur dioxide (see diagram). The reaction is therefore much faster.
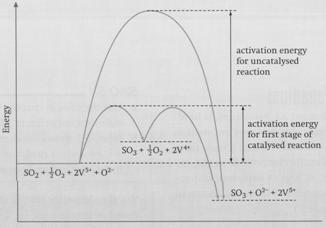
The
course of reaction involving V2O5 as a catalyst
Chromium
Element 24, chromium, is
hard but brittle, and has an attractive silvery appearance which has led to it
coating the bumpers and other bits of bicycles and cars.
Chromium forms compounds in the +2, +3, +4, +5 and +6 oxidation states, out of which +3 and +6 are the two most often seen in chromium compounds.
An oxyanion is a negatively charged ion containing oxygen. In the +6 state chromium forms two different oxyanions. These are the orange dichromate(VI) ion, Cr2O72- and the yellow chromate(VI) ion, CrO42-. They are commonly used as potassium salts, potassium dichromate(VI), K2Cr2O7, and potassium chromate(VI), K2CrO4. In solution each of these oxyanions can be converted into the other by altering the pH, since the following equilibrium exists:
Cr2O72- + H2O
![]() 2CrO42- + 2H+
2CrO42- + 2H+
Cr2O72-
is formed in acidic solution, while CrO42- the ion formed
in alkaline solution (le Chatelier).
Note: The interconversion of Cr2O72- and CrO42- can be shown as an equilibrium in three different ways:
Cr2O72- + H2O
![]() 2CrO42- + 2H+
2CrO42- + 2H+
Cr2O72- + ![]() 2CrO42-
+ H+
2CrO42-
+ H+
Cr2O72-
+ 2OH- ![]() 2 CrO42-
+ H2O
2 CrO42-
+ H2O
All three versions are valid and predict the same outcome: CrO42- is formed in alkaline solution, Cr2O72- in acidic solution.
Potassium dichromate(VI) in acid solution is a strong oxidising agent, being itself reduced to Cr3+ ions which are very stable:
Cr2O72- + 14H+ ![]() 2Cr3+ + 7H2O Eø = +1.33 V
2Cr3+ + 7H2O Eø = +1.33 V
In aqueous solution Cr3+
forms the violet [Cr(H2O)6]3+ complex ion.
However, when potassium dichromate(VI) in acid solution is used as an oxidising
agent, the colour change observed is from orange to green. Particularly when
hot, [Cr(H2O)6]3+ undergoes ligand
substitution reactions forming green species. This is the reason why most solutions
and solids containing Cr3+ are green. For example, in hydrochloric
acid the dark-green complex ion [Cr(H2O)4Cl2]+
is formed.
Uses of chromium
In 1913, Harry Brearley, a
After further investigation and scientific fiddling around, Brearley settled on an alloy consisting of 70% iron, 20% chromium and 10% nickel as the ideal 'stainless steel'. The chromium and nickel in the alloy quickly form a layer composed of their oxides on the surface of the steel when it first comes in contact with the air. Unlike iron oxide, which is flaky and porous, this oxide layer is tenacious and impervious to air and water - it protects the steel underneath.
If smaller amounts of chromium are added to steel, the resulting alloy is very hard. Steel hardened in this way is used for making many tools.
Cobalt
Element 27, cobalt, is a
hard, unreactive, white metal with a slightly blue appearance. It forms a wide
range of compounds in the +2 and +3 oxidation states, of which the +2 state is
usually the most stable. Co2+ forms the stable [Co(H2O)6]2+
ion, which is pink - as are most octahedral Co2+ complexes.
Tetrahedral complexes of Co2+, such as [CoCl4]2-,
are mostly blue.
Cobalt chloride paper is
used as a test for water. When dry, the paper is blue which is the colour of
anhydrous cobalt chloride, CoCl2. Following the addition of water,
the [Co(H2O)6]2+ ion is formed and the paper
turns pink.
Co3+ forms the aqueous complex [Co(H2O)6]3+ which is blue. However, it is so easily reduced to [Co(H2O)6]2+ that there is no simple aqueous chemistry involving the [Co(H2O)6]3+ ion. There are, in fact, very few simple compounds containing Co3+, and those that do exist, such as CoF3, instantly react with water to give a Co2+ compound. The water is oxidised in the reaction, producing oxygen gas. In complexes with ligands other than water, Co3+ is stable, however, and this leads to the 'wide range' of compounds already referred to, exhibiting an equally wide range of colours.
[Co(H2O)6]2+
is a good example of a complex ion that changes colour due to ligand substitution.
The endothermic change
[Co(H2O)6]2+
+ 4Cl- ![]() [CoCl4]2- + 6H2O
[CoCl4]2- + 6H2O
takes place in CoCl2(aq) if either a high concentration of CI- or a high temperature is applied.
An example of the enhanced
stability of Co3+ with different ligands is the stability of [Co(NH3)6]3+.
Compare the Eø values:
[Co(H2O)6]3+ + e- ![]() [Co(H2O)6]2+
; Eø = +1.81 V
[Co(H2O)6]2+
; Eø = +1.81 V
[Co(NH3)6]3+
+ e- ![]() [Co(NH3)6]2+
; Eø = +0.11 V
[Co(NH3)6]2+
; Eø = +0.11 V
(The magnitude of the
standard electrode potential, Eø, is an indication of the
reactivity. The larger the magnitude, the more reactive.)
A wide variety of reducing agents can reduce [Co(H2O)6]3+ to [Co(H2O)6]2+, including water, iodide ions and silver metal. The reduction of [Co(NH3)6]3+ to [Co(NH3)6]2+ requires much stronger reducing agents, however, like iron metal or V2+ ions. The enhanced stability of [Co(NH3)6]3+ is due to ammonia being a stronger ligand than water i.e. ammonia forms stronger dative bonds.
Copper
Element 29, copper, is a
reddish-gold colour. It is soft and ductile, and its ability to conduct heat
and electricity is second only to silver. It was first smelted from its ores
over 5500 years ago. Early metallurgists in
Copper forms a wide range
of stable compounds in the +2 oxidation state but, unlike most other transition
metals, copper also forms compounds in the +1 oxidation state. While compounds
and complexes containing Cu2+ are virtually all green or blue, compounds
containing Cu+ are white solids.
Aqueous solutions containing Cu2+ ions are stable; however, aqueous solutions containing Cu+ ions are not. They disproportionate to give Cu atoms and Cu2+ ions:
2Cu+ ![]() Cu
+ Cu2+
Cu
+ Cu2+
The disproportionation of
Cu+ ions only takes place in aqueous solution. Dry copper(I)
compounds and those that are insoluble in water are therefore stable.
All the copper(I) halides
are extremely insoluble in water. If a solution containing Cu2+ ions
is mixed with a solution containing an excess of iodide ions, the iodide ions
will reduce the Cu2+ ions to Cu+ ions. The iodide ions
are oxidised to iodine.
Cu2+ + I-
![]() Cu+
+ ½ I2
Cu+
+ ½ I2
The Cu+ ions
then combine with more iodide ions to form a precipitate of copper(I) iodide:
Cu+ + I- ![]() CuI
CuI
The overall equation is therefore:
Cu2+(aq) + 2I- ![]() CuI(s) + ½I2(aq)
CuI(s) + ½I2(aq)
Although the CuI
precipitate that forms is actually white, it always appears a brown colour due
to the iodine that forms at the same time. The Cu+ ions in CuI are
stable and do not disproportionate because they are not in aqueous solution.
Copper alloys
Copper is a constituent of
many useful alloys.
Copper and zinc make
brass. Its resistance to corrosion, pleasant ringing tones and attractive
gold-coloured appearance all explain why brass is used to make musical
instruments. Brass is harder than copper. Both metals are used in central
heating systems. Being more ductile, copper is drawn out to make the pipes:
Being harder, brass is more suitable for the joints, which are made by casting
or machining.
Copper and tin make
bronze. Bronze is harder than copper and is used to make statues and medals.
Bronze (in this case 97%
copper, 2.5% zinc and 0.5% tin) is used for 1p and 2p coins. However, since
September 1992, the price of copper has made it too expensive to make 1p and 2p
coins out of bronze, so steel coated in a thin layer of copper has been used
instead - except for some of the 2p coins issued in 1998. You can distinguish
the copper-plated steel coins from the older ones with a magnet.
Cupro-nickel is one of the
alloys used for
Analysing the amount of copper in an alloy
If a piece of brass is dissolved in nitric acid, the solution obtained is a mixture of copper(II) nitrate and zinc nitrate. Analysing this mixture can tell us how much copper it contains. If the mass of the original piece of brass is also known, then the composition of the original alloy can be calculated.
The analysis is done by
adding an excess of KI(aq) to the mixture containing Cu2+. As has
already been seen, this causes the precipitation of all the copper as copper(I)
iodide:
2Cu2+ + 4I- ![]() 2CuI
+ I2
2CuI
+ I2
The amount of iodine
produced can then be found by titrating the resulting mixture with a sodium
thiosulphate (Na2S2O3) solution of known
concentration. The thiosulphate ions reduce the iodine back to iodide ions:
2S2O32- + I2 ![]() S4O62- + 2I-
S4O62- + 2I-
As the end-point of this
titration is neared, the brown colour due to the iodine becomes faint. Adding a
little starch solution produces a dark blue colour, which in turn disappears
when enough sodium thiosulphate solution has been added to react with all the
iodine. The disappearance of a dark blue colour is much easier to judge
accurately than the disappearance of a pale brown colour.
The two equations above
show us that two Cu2+ ions produce one iodine molecule, which then
reacts in the titration with two thiosulphate ions. The number of moles of
thiosulphate added in the titration is therefore the same as the number of moles
of copper originally present. From this the original mass of copper in the
alloy can be calculated. This measurement is known as 'estimating the
composition of the alloy'. The word 'estimating' in this context does not suggest
inaccuracy!
Precipitating
transition metal hydroxides
When sodium hydroxide is added to a solution of a transition metal, a precipitate of the metal hydroxide always appears.
Let us consider why this happens. When a metal aqua ion is placed in water, an equilibrium is established:
[M(H2O)6]n+ ![]() [M(H2O)5(OH)](n-1)+ + H+
[M(H2O)5(OH)](n-1)+ + H+
This reaction is called a
‘hydrolysis’ because a water molecule has undergone a reaction in which it has
split into
When alkali is added to a solution of a transition metal, they attack the strongest acid present, i.e. H+:
![]() H2O
H2O
This upsets the hydrolysis equilibrium and when the equilibrium has been completely pushed over to the right hand side, a new equilibrium is established:
[M(H2O)5(OH)](n-1)+ ![]() [M(H2O)4(OH)2](n-2)+ + H+
[M(H2O)4(OH)2](n-2)+ + H+
For example, when sodium hydroxide is added to copper(II) sulphate solution, a gelatinous (jelly-like) blue precipitate appears. The hydroxide ions firstly upset the hydrolysis reaction:
[Cu(H2O)6]2+ ![]() [Cu(H2O)5(OH)]+ + H+
[Cu(H2O)5(OH)]+ + H+
When this equilibrium is pushed fully to the right hand side by addition of excess hydroxide ions, the second equilibrium is established:
[Cu(H2O)5(OH)]+ ![]() Cu(H2O)4(OH)2 + H+
Cu(H2O)4(OH)2 + H+
The product, copper(II) hydroxide, Cu(H2O)4(OH)2, is uncharged. In this case, there is insufficient solvation energy to overcome the lattice enthalpy, so a solid (i.e. precipitate) is formed. Usually, copper(II) hydroxide is written in its simpler form, Cu(OH)2. However, it should be remembered that Cu(H2O)4(OH)2 and Cu(OH)2 represent the same thing. The co-ordinated water molecules are present in both.
A similar sequence occurs whatever the metal ion so that we can summarise the reactions of sodium hydroxide with metal ions:
Cu2+(aq) + 2OH-(aq) ![]() Cu(OH)2(s) pale (
Cu(OH)2(s) pale (
Fe2+(aq) + 2OH-(aq) ![]() Fe(OH)2(s) green gelatinous precipitate
Fe(OH)2(s) green gelatinous precipitate
Fe3+(aq) + 3OH-(aq) ![]() Fe(OH)3(s) brown gelatinous precipitate
Fe(OH)3(s) brown gelatinous precipitate
Mn2+(aq) + 2OH-(aq) ![]() Mn(OH)2(s) cream gelatinous precipitate
Mn(OH)2(s) cream gelatinous precipitate
Cr3+(aq) + 3OH-(aq) ![]() Cr(OH)3(s) grey-green gelatinous precipitate
Cr(OH)3(s) grey-green gelatinous precipitate
Co2+(aq) + 2OH-(aq) ![]() Co(OH)2(s) blue-green gelatinous precipitate
Co(OH)2(s) blue-green gelatinous precipitate
Note: (i) Mn+(aq) is a shorthand for
[M(H2O)6]n+
(ii) Fe(OH)3 is unstable and rapidly dehydrates to form rust (Fe2O3).
(iii) These precipitates will form with any source of hydroxide ion. For example, addition of dilute aqueous ammonia to copper(II) sulphate solution will give a gelatinous blue solution.